|
Abstract: We present a program for the prediction of biological activity spectra for drug-like organic substances. New lead compounds can be found on the basis of predicted biological activity spectra. In house and Internet versions of the PASS program are discussed. Keywords: biological activity spectrum, computer-aided prediction, computer system PASS (Prediction of Activity Spectra for Substances), applications in computer-aided drug discovery, prediction via Internet. Most of known biologically active substances have many different biological activities that cause both main (therapeutic) and supplementary (side) actions. Some of these activities are found during the initial preclinical study; others are found unfortunately too late in clinical trials (see, for example, the Fluorouracil's activities in Table 1). Sometimes, many years after the first launch of a drug additional activities are discovered that become the base for a new therapeutic application (see some examples in Table 2). Most computer-aided drug-discovery methods are used to study a single, or only a few activities of a compound class. [1-5]. A program that predicts simultaneously pharmacological effects, mechanisms, and specific toxicities on the basis of the 2D chemical structure is the tool of choice to get an early indication if a compound could be a potential lead. Victor Avidon proposed this idea more than 35 years ago [6, 7]. In the framework of national registration system of the UDSSR this technology has been formerly developed and tested on new chemical compounds synthesized in the USSR [8, 9]. The program was revised several times. The theoretical analysis went through several approaches and the accumulated experience of finding new leads allows constant improvements [10-14].
|
The PASS team is permanently collecting and evaluating the information about new pharmaceutical substances and lead compounds, to update the PASS training set and extend PASS predictive abilities on new chemical classes and novel biological activities:
, Figure 1. Increase of the number of
compounds over years that are abstracted for the knowledge base
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

Figure 2. Increase of the number of predictable activities over years The current version of PASS predicts ca. to 4130 pharmacological effects, mechanisms of action, and other effects, see Table 1. [15]. We provide a list of activities. In the following we show the methods used in PASS, examples of practical applications, and how you can evaluate PASS yourself by using it on line as demo version, or as evaluation version.
|
Table 1. List of biological effects
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Presentation of biological activities in PASS Let's define biological activity as the result of a compound's interaction with an biological entity. In clinical studies the entity is the human organism. In preclinical testing it can be animals (in vivo) or experimental models (in vitro). The biological activity depends on a compound's structure, charge distribution, physico-chemical properties, and more. The activity depends on the biological entity (species, sex, age, etc.), on the mode of treatment (dose, route), etc. Any biologically active compound reveals a wide spectrum of different effects. Some of them are useful in treatment of diseases but others cause various side and toxic effects. All activities caused by the compound are considered to be the "biological activity spectrum of the substance". If the experimental conditions can not be defined narrowly, i.e if difference in species, sex, age, dose, route, etc. is neglected the biological activity can be identified only qualitatively (“yes’/“none”, “active”/“inactive”). Thus, the "biological activity spectrum" is defined as an "intrinsic" property of a compound depending only on its structure and physico-chemical characteristics. Qualitative presentation allows integrating information concerning biologically active compounds collected from many different sources into the general PASS training set. Any property of chemical compounds, which is determined by their structural peculiarities, can be used for prediction by PASS. It was shown, that the applicability of PASS is broader than the prediction of biological activities. For instance, this approach was successfully used for prediction of such general property of organic molecules as drug-likeness (Anzali et al., 2001). Chemical structure description in PASS. The 2D structural formulae of compounds were chosen as the basis for description of chemical structure because this is the only information available at the early stage of research. Thus, using the structural formula as an input data, one can obtain the estimates of biological activity profiles even for virtual molecules, prior to their chemical synthesis and biological testing. Many different characteristics of chemical compounds can be calculated on the basis of structural formulae. In the earliest versions of PASS (Poroikov et al., 1993; Filimonov et al., 1995; Filimonov and Poroikov, 1996) we used the Substructure Superposition Fragment Notation (SSFN) codes (Avidon et al., 1982). However, SSFN, like many other structural descriptors, reflects rather abstraction of chemical structure by the human than the nature of ligand-target interactions, which are the molecular mechanisms of biological activities.
|
The Multilevel Neighbourhoods of Atoms (MNA) descriptors (Filimonov et al., 1999) have certain advantages in comparison with SSFN. These descriptors are based on the molecular structure representation, which includes the hydrogen atoms according to the valences and partial charges of other atoms and does not specify the types of bonds. MNA descriptors are generated as recursively defined sequence:
The mark of atom may include not only the atomic type but also any additional information about the atom. In particular, if the atom is not included into the ring, it is marked by “-”. The neighbor descriptors D1D2....Di are arranged in unique lexicographic order. Iterative process of MNA descriptors generation can be continued covering first, second, etc. neighborhoods of each atom. The molecular structure is represented in PASS by the set of unique MNA descriptors of the 1st and 2nd levels (Figure 3). The substances are considered to be equivalent in PASS if they have the same set of MNA descriptors. Since MNA descriptors do not represent the stereochemical peculiarities of a molecule, the substances whose structures differ only stereochemically, are formally considered as equivalent.
Figure 3. Structural formula of nicotinic acid and its MNA descriptors of the 1st (left column) and 2nd (right column) levels New QNA (Quantitative Neighbourhoods of Atoms) descriptors were recently developed, which allow the analysis of quantitative structure-activity relationships (Filimonov et al., 2009).
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Mathematical Approach PASS algorithm of biological activity spectrum prediction is based on Bayesian estimates of probabilities of molecule’s belonging to the classes of active and inactive compounds, respectively. The mathematical method is described in several publications (Lagunin et al., 2000; Stepanchikova et al., 2003; Poroikov and Filimonov, 2005; Filimonov and Poroikov, 2006; Filimonov and Poroikov, 2008), and its details will not be discussed here. Only general description necessary for interpretation of prediction results is presented below. Since the main purpose of PASS is the prediction of activity spectra for new molecule, the general principle of the PASS algorithm is the exclusion from SAR Base the substances, which are equivalent to the substance under prediction. The structural formula of molecule, for which PASS prediction should be carried out, is presented as a MOL file (for the set of molecules – as SDFile). The predicted activity spectrum is presented in PASS by the list of activities with probabilities "to be active" Pa and "to be inactive" Pi calculated for each activity (Figure xxx). The list is arranged in descending order of Pa-Pi; thus, the more probable activities appeared at the top of the list. Only activities with Pa>Pi are considered as possible for a particular compound. The list can be shortened at any desirable cutoff value, but Pa>Pi is used by default. If the user chooses rather high value of Pa as a cutoff for selection of probable activities, the chance to confirm the predicted activities by the experiment is high too, but many existing activities will be lost. For instance, if Pa>90% is used as a cutoff, about 90% of real activities will be lost; for Pa>80%, the portion of lost activities is 80%, etc. It is necessary to keep in mind that probability Pa reflects the similarity of molecule under prediction with the structures of molecules, which are the most typical in a sub-set of “actives” in the training set. Therefore, usually there is no direct correlation between the Pa values and quantitative characteristics of activities. Even active and potent compound, whose structure does not resemble the typical structures of “actives” from the training set, may obtain a low Pa value during the prediction (even negative Pa-Pi values could be observed). This may be explained by the way how the appropriate estimates are constructed: the values Pa for “actives” and Pi for “inactives” are distributed uniformly. |
Taking this into account, the following interpretation of prediction results is possible. If, for instance, Pa=0.9, then for 90% of “actives” from the training set the appropriate estimates are less than for this compound, and only for 10% of “actives” these values are higher. If one declines the suggestion that this compound is active, he will make a wrong decision with probability 0.1. In case if Pa<0.5, but Pa>Pi, for more than half of “actives” from the training set the appropriate estimates are higher than for this compound. If one declines the suggestion that this compound is active, he will make a wrong decision with probability less than 0.5. In such case the probability to confirm this kind of activity in the experiment is small, but if it will be confirmed, more than 50% chances that this structure has a high novelty and may become NCE. If the predicted biological activity spectrum is wide, the structure of the compound is quite simple, and does not contain peculiarities, which are responsible for the selectivity of its biological action. If it appears that the structure under prediction contains several new MNA descriptors (in comparison with the descriptors from the compounds of the training set), then the structure has low similarity with any structure from the training set, and the results of prediction should be considered as rather rough estimates. Based on these criteria, one may choose which activities have to be tested for the studied compounds on the basis of compromise between the novelty of expected pharmacological action and the risk to obtain the negative result in experimental testing. Certainly, one could also take into account a particular interest to some kinds of activity, experimental facilities, etc. We have developed a special application CWM Lead Finder which matches with clustering algorithms the biological spectra of a set of compounds with known biological activity and a set of untested compounds.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
The accuracy and efficiency of more than 200 various mathematical approaches were tested to select the most relevant algorithms [16]. One of the methods that provides a satisfactory quality of prediction is described below in more details. Definitions: n is the total number of compounds in the training set; pj = nj/n is the estimate of a priori probability of activity
j; Prj is the initial estimate of the probability of the
activity j for the compound under prediction; The 1st kind error is observed when the compound under
prediction actually is active but Prj < CPj; LOO is the leave-one-out procedure. For each compound in the training set the values n, ni, nj, nij are changed to n-1, ni-1, and nj-1, nij-1 when it has activity j, and the estimates Prj are calculated. MEP is the maximal error of prediction (see below). --->
|
Algorithm of Prediction Structural descriptors are generated for the compound under prediction. The following values are calculated for each activity: uj = SiArcSin{ri(2pij-1)}, vj = SiArcSin{ri(2pj-1)} sj = Sin(uj/m), tj = Sin(vj/m) Prj = (1+(sj-tj)/(1-sjtj))/2 Validation criteria: The LOO estimates of
Prj are calculated for each compound in the training set. E1j(CPj*) = E2j(CPj*) are calculated. The maximal error of prediction MEP is: MEPj = E1j(CPj*) = E2j(CPj*) Results of the prediction: The probability to be active is: Pa = E1j(Prj) The probability to be inactive is: Pi = E2j(Prj) The result for the prediction is presented as the list of activities with appropriate Pa and Pi, sorted in descending order of the difference (Pa-Pi)>0.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
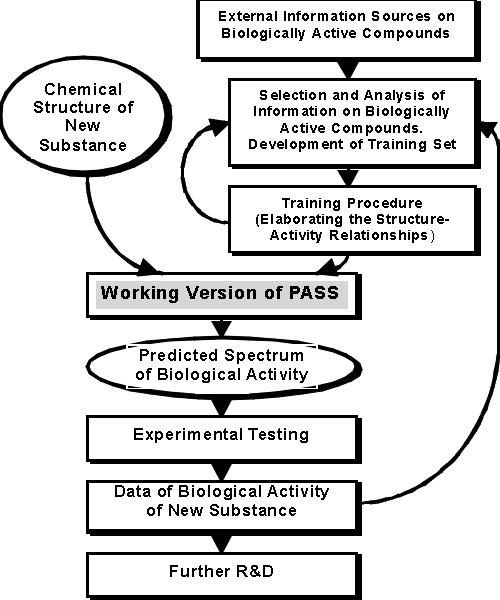
Figure 1. The Process of PASS Development
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
The principal elements of PASS include the Training Set, Chemical Structure Description, Biological Activity Description, and the Mathematical Approach. They are described in more details below. PASS 1.611 training set consists of about 46'000 of biologically active compounds, from which about 15,000 substances are already launched drugs, and about 13,000 drug-candidates are under clinical or advanced preclinical testing, now. Since 1972 this training set is compiled from many sources including: ---> |
publications, patents, databases, private communications, etc. For the majority of compounds, included into the training set, the biological activity spectrum of each compound was studied in detail. In PASS Pro the customer can create easily his own training set. A training set consists of a SDFile with the field activity_prediction. This file is read into PASS. It takes about 5 minutes to read a training set of 1000 compounds. Chemical Structure Description The structure of a compound is described by descriptors. We call them Multilevel Neighborhoods of Atoms (MNA). The general idea is illustrated below for Ethanol. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
At the first step, the 1st and 2nd levels neighborhoods for atoms are generated, see above. At the second step, the duplicate MNA descriptors are eliminated. H(C(HHHC)) Despite the possibility to continue the procedure calculating the 3rd, 4th, etc. levels of atoms' neighborhoods, only descriptors of the 1st and 2nd levels are used, because this approximation is shown to provide the best quality of prediction. Biological Activity Description Let's define biological activity as the result of a compound's interaction with an biological entity. In clinical studies the entity is the human organism. In preclinical testing it can be animals (in vivo) or experimental models (in vitro). The biological activity depends on a compound's structure, charge distribution, ---> |
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
The quality of prediction can be calculated by leave-one-out cross validation. Each of the compounds is subsequently removed from the training set and the prediction of its activity spectrum is carried out on the basis of the remaining part of the training set. The result is compared to the known activity of the compound, and the maximal error of prediction (MEP) is calculated, and averaged over all compounds and activities. This error is about 0.15 for the current version of PASS. The average accuracy of prediction using LOO cross-validation is about 0.85. Such accuracy is enough for practical use. Especially taking into account that random guess would produce a number of 1/1000 = 0.001 for 100 activities. Interpretation of the Prediction's Results The total number of MNA descriptions of the compound, and the number of new MNA descriptors are shown with the result. If the number of new descriptors is more than 3, the result of the prediction may questionable.---> |
Pa and Pi are the estimates expressed as probability of the compound to be active and inactive. The values vary from 0 to 1. Only activities with Pa > Pi are considered as possible for a particular compound. If Pa > 0.7. the chance to find the activity experimentally is high. But, in many cases the compound may occur to be a close analogue of known pharmaceutical agents. If 0.5 < Pa < 0.7 the chance to find the activity experimentally is less, but the compound is probably not so similar to known pharmaceutical agents. If Pa < 0.5 the chance to find the activity experimentally is even less. But, the chance increases of finding a structurally new compound. Thus, one may choose which activities have to be tested in ones compounds on the basis of compromise between expected novelty of pharmacological agent and risk to get too many false positives. Using a biological profile = several important activities one can increase the quality of prediction considerably. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
It takes about 30 minutes to calculated the biological activity spectra for 100'000 compounds on an ordinary IBM PC Pentium/500 MHz. One can effectively use PASS for predicting activities of many compounds from large in-house and commercial databases. At NCI the PASS parameters are provided with each compound, by updating the database periodically. One also can calculated the PASS parameters on the fly. This would have the advantage that one gets also a prediction for compounds that one searches on-line at CAS, or at DiscoveryGate. PASS can be useful applied for:---> |
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
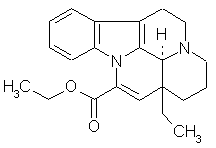
Revealing New Effects and Mechanisms of Action This is considered below on the example of predicting the biological activity spectrum for the well-known cerebrotonic drug Cavinton (Vinpocetin). This was launched by Gedeon Richter (Hungary) more than twenty years ago. Its structural formula and predicted biological activity spectrum are given below. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Predicted biological activity spectrum for Cavinton 45 Descriptors, 0 New Descriptors, 47 Predicted Activities
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Cavinton is used in medicinal practice for twenty years. Many activities that were found in preclinical testing and clinical trials during this period are compared with the result of the prediction. According to the available literature only 16 of 47 predicted activities of Cavinton are already found. These activities are marked by "+" in the Table above. In particular, ASS predicts the vasodilator and spasmolytic activities (Pa=0.855 and 0.540). It corresponds with the well-known pharmacological effects of Cavinton. It causes vasodilatation, increases the brain blood flow and metabolism. Antihypoxic and Antiischemic effects are also predicted for Cavinton (Pa=0.700 and 0.656 respectively). Cavinton is used for these purposes. Cavinton is predicted as Lipid peroxidase inhibitor (Pa=0.650), agent for cognition disorders treatment (0.648), agent for acute neurological disorders treatment (0.577), etc. Cavinton has all these activities.---> |
The predicted biological activity spectrum of Cavinton suggests several new application of the substance. Among them are: Multiple sclerosis treatment (Pa=0.900); Antineoplastic enhancer (0.812), Antineoplastic Alkaloid (0.225) and Antitumor-Cytostatic (0.236); Antiparkinsonian rigidity-relieving (0.271) and Antiparkinsonian tremor-relieving (0.243); etc. While the Multiple sclerosis treatment is predicted with high probability, all other additionally predicted activities have relatively small values of Pa. Similarly, the predicted activity spectrum for any compound provides ideas for further testing. As a result some new effects and mechanisms will be found for old substances. Varying the cutoff value of Pa one may choose the desirable level of novelty vs. acceptable risk of negative result. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
A researcher can define desirable and not desirable activities for a compound. She can select such compounds from a set of structures with the help of PharmaExpert. For example, among the 15630 compounds of the ChemStar database (http://www.chemstar-ru.com) 959 compounds are predicted as Endothelin antagonist, 236 compounds as Angiotensin II antagonist, 57 compounds as Angiotensin converting enzyme inhibitor. ---> |
If the purpose of the study is to find the compounds with dual mechanism of Antihypertensive effect, e.g. Angiotensin converting enzyme inhibitor + Endothelin antagonist, only 11 compounds are predicted as having both activities. The best from the hits has Pa =0.170 (Endothelin antagonist) and Pa=0.244 (Angiotensin converting enzyme inhibitor). Based on this result one may decide either to test these 11 compounds or to carry out the prediction and selection for compounds from another database. In any case varying the cutoff value of Pa it is possible to choose the compounds with less or higher novelty (see: Interpretation of the Prediction's Results). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Selecting the Most Prospective Compounds for Highthroughput Screening. Sometimes one is interested in activities that are not yet included in PASS, and the data are not available to train ones own knowledge base for PASS Pro. In such cases two other strategies are suitable. The first strategy is based on the hypothesis that the more
activities are predicted for a compound, the higher is the chance to find
any useful pharmacological action for this compound. For each compound the following value is
calculated: P = [S Pa/(Pa+Pi )]/n All compounds are arranged in the descending order of P values, and only compounds with the highest values of P are selected for screening. The second strategy is based on the hypothesis that the more "novel" a compounds is, the higher is the probability to find a NCE. Thus, the compounds with the highest amount of new descriptors are selected. Both strategies were tested on datasets including 10,000 - 70,000 compounds and their efficacy is shown [31].---> |
Determining Relevant Screens for a Particular Compound. Testing can be organized in descending order of difference (Pa-Pi) for different activities. For example, if we consider the example of Cavinton, it should be studied in the following tests: Peripheral vasodilator (0.929-0.004), Multiple sclerosis treatment (0.900-0.000), Vasodilator (0.855-0.005), Abortion inducer (0.844-0.003), Antineoplastic enhancer (0.812-0.001), Coronary vasodilator (0.760-0.006), etc. In this case both safety and efficacy of a new compound will be characterized more comprehensively. Moreover, it is shown that the economic viability of such approach to testing is more than 500% [32]. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
The predictions of PASS were confirmed by experiment. Some of these examples are given below. The activity spectra have been predicted for 300 new chemical compounds, synthesized in the Chemical-Pharmaceutical Research Institute (Novokuznetzk). Twenty compounds have been selected for testing as probable antiulcer agents. Nine compounds have been synthesized and tested. A potent antiulzer activity was found for 5 of these compounds. These new antiulcer agents are NCE [33]. The economic advantage is about (300/20)100 = 1500% in this study. The activity spectra have been predicted for 520 new chemical compounds, synthesized in the Institute of Organic Chemistry of Russian Academy of Science (Moscow). Fourteen compounds have been selected for testing as the most prospective. It was shown that the results of 22 experiments made on 5 various kinds of activity, coincide with predictions in 20 cases. The accuracy of prediction is about 90%. Based on the predicted biological activity spectra for about 20 macroheterocyclic compounds, 2 antitumor leads were found.[34]. New antibacterial agents were found based on the biological activity spectra for derivatives of 1-amino-4-(5-arylozaxolyl-2)-butadiens-1,3 [35]. Analgesic, antiinflammatory, antioxidant and some additional activities were predicted and confirmed by experiment for some thiazole derivatives [36]. ---> |
These and some other examples demonstrate that the approach to predicting many biological activities simultaneously can be effectively applied to compounds from different chemical series to find various pharmacological actions. Naturally, the PASS approach has some limitations. They are:
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Since July 1998 PASS is open for free testing via Internet (http://www.ibmh.msk.su/PASS/default.htm). Anyone who would like to obtain additional information about the biological potential of her compound may fill the registration form and send the structure file in ISIS (MDL Information Systems, Inc.) "MOLl" format. Such files can be prepared, for example, with the chemical editor ISIS/Draw (MDL Information Systems, Inc.). ISIS/Draw is available free for personal or non-commercial use from the MDL web site http://www.mdl.com. The molfile can be prepared with ISIS/Draw by drawing the sucture using the menu options and the mouse. After that one can choose "Edit" a "Select All". When the molecule is selected as a total, choose "File" a "Export" a "Molfile". Files have to be saved on the disk under a name defined by the user.
|
When the molfile is prepared and the registration form is filled in the Internet version of PASS, one may click on the option "Browse" and select the molfile, and click on "Submit now" and wait for the result. In case of any problem, please send an E-mail to pass@ibmh.msk.su. A new program is developed for predicting biological activity of drug-like compound on the basis of the 2D structure. It can be applied effectively in finding new leads. This is demonstrated on examples of both compounds with known activities and new synthesized structures studied as potential pharmacological agents. The program can be tested easily via the Internet, by downloading a light version, or by applying for the evaluation of the full version. Please send an email to us Acknowledgments We gratefully acknowledge MDL Information Systems, Inc. for providing ISIS/Host, ISIS/Base and the MDDR database used in this study. This is an edited version of the original paper, A. Kos 3.2.03. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
FLUOROURACIL'S KNOWN ACTIVITIES (CAS NO. 51-21-8)
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
EXAMPLES OF PHARMACEUTICALS, WHICH ADDITIONALLY DISCOVERED
ACTIVITY
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||